BIOSCIENCE 遺伝子改変マウス関連受託
遺伝子改変動物作製
ゲノム編集技術
ゲノム編集技術(ZFN, TALEN, CRISPR/Casシステム等)は、受精卵にRNAをインジェクションすることで遺伝子改変動物が作製できるという共通点があります。標的とする領域でヌクレアーゼ活性を持つ酵素が働きDouble Strand Break (DSB)を起こさせます。DSBが起こると、NHEJ(non-homologous end-joining:非相同末端結合)か、HR(homologous recombination:相同組換え)の2種類のいずれかにより修復が起こります。NHEJはDSBが起こった部位を単純に結合させるというもので、DSB発生時のメインの修復機構となります。しかし、連結時に数塩基~数十塩基のinsertion(挿入) あるいは deletion(欠失)変異(indel変異)が生じることがあります。この修復エラーを利用してframe-shift変異を導入し、標的遺伝子のノックアウトが可能になります。HRによる修復では修復に際して、鋳型となる正しい配列が存在するため、NHEJと違い正確な修復が可能です。通常、HRによる修復は姉妹染色分体を鋳型として行われますが、標的とするDSB導入部位近辺の配列(相同領域)を含んだドナーDNAをRNAと一緒にインジェクションすることで、ドナーDNAを鋳型としてHRによる修復を起こすことができます。その際に、ドナーDNAにpoint mutationを入れておくことで、ゲノム中に正確にpoint mutationを導入し、モデル動物を作製することも可能です。また、HRを用いることにより、loxP配列や大きなサイズのGFP(Green Fluorescent Protein)遺伝子の導入もできます(図1)。
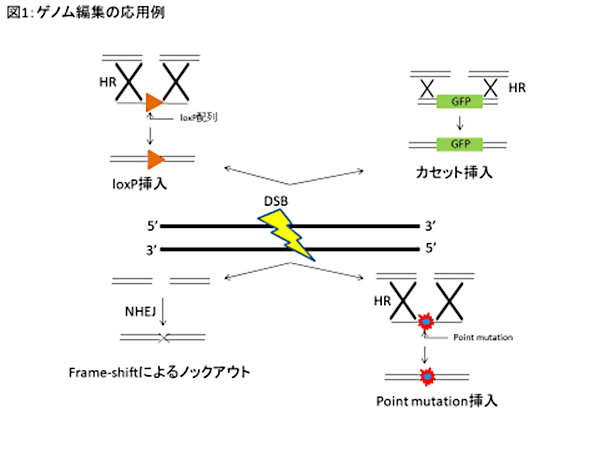
この技術により、ES細胞の樹立が困難であった生物でも遺伝子改変生物作製の報告が次々となされています。また、遺伝子改変動物作製のみにとどまらず、遺伝子治療に至るまで、様々な活用法が期待されています。以下に3種類のゲノム編集技術について、それぞれの特徴を紹介します(図2)。
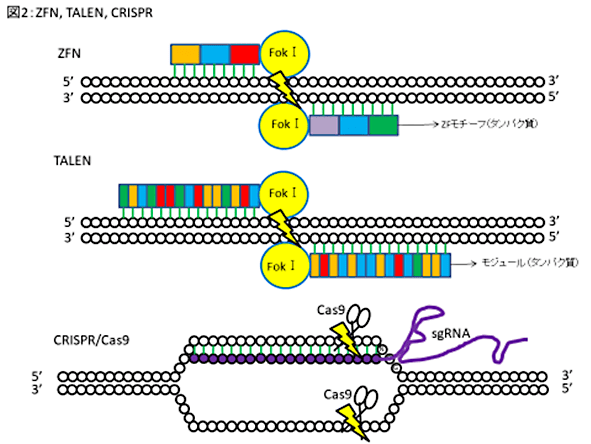
- ZFN
- ZFNはゲノム編集技術の第1世代です。3塩基を認識するZFモチーフとその末端にFokⅠヌクレアーゼを結合させた人工のキメラタンパク質が、標的領域でヘテロダイマーを形成して標的DNA部位でDSBを起こすことが1996年に報告されました。一つのZFユニットが3塩基を認識し、3~6ユニットの組合せで9~18塩基を認識することになります。5~6塩基対のスペーサー領域を挟む形でセンス鎖とアンチセンス鎖に設計することにより標的とする配列を認識させます(図2)。
- TALEN
- ZFNのDNA結合ドメインを植物病原細菌であるXanthomonas属のもつTALエフェクターに変えた第2世代のTALENの開発が2011年に報告されました。TALエフェクターがDNAに結合する際は、1つのモジュールが1つの塩基を認識します。TALENもZFNと同じくスペーサーを挟むようにセンス鎖(15塩基~20塩基を認識)とアンチセンス鎖(15塩基~20塩基を認識)をそれぞれ作製します。
- CRISPR/Casシステム
- 2013年には原核生物の獲得免疫として働くCRISPR/Casシステムを利用した第3世代のゲノム編集ツールが開発されました。CRISPR/Casシステムはファージ感染や接合などを介して侵入する外来DNAを断片化して内在性のゲノム中に取り込みます。再び侵入してきた際に、1回目に取り込んだDNAの転写の結果できるRNAを用いて外来DNAを認識し、CASヌクレアーゼにより切断します。ZFNおよびTALENとは違い、標的とするDNAをRNAが認識します。すなわち、標的配列と相補的配列を持つguide RNAとヌクレアーゼ活性を持つCASタンパク質を準備するだけでいいのです。ZFNやTALENではDNA結合ドメインと切断ドメインを融合タンパク質として作製する必要があり、複雑なベクター構築が必要でしたが、CRISPR/Casシステムでは標的配列に対応する塩基を変えるだけでより簡便にツールを構築できます。ただし、認識塩基の近傍にPAM(protospacer adjacent motif)と呼ばれる数塩基が必要なため、標的配列決定の自由度を下げますが、ゲノム編集活性はZFNやTALENに比べて高くなっています。現在、CRISPR-Cas9やCRISPR-Cas12a等多くの種類が発見されています。手軽でありながらゲノム編集活性が高く、低コストで編集ツールが準備できることから、培養細胞、マウス、ラット、ブタからサルに至るまで、広く応用されています。
課題
このように汎用性の高い方法にもいくつか課題が残されています。以下にその課題とそれに対する対応策の一部を紹介します。
- 課題1. モザイク
- ゲノム編集技術を用いた遺伝子改変動物作製の際、前核期胚にRNAをインジェクションしますが、受精卵の発生が進み、2細胞期胚や4細胞期胚でヌクレアーゼによる標的配列の切断が起こると、様々な変異を持った細胞が同居するモザイク個体が生まれてくることがあります。対策として、単純なノックアウト動物作製の場合は、F0で詳細な検査を実施するよりF0個体と野生型個体を交配させ、目的変異を持つF1を作製した上で、遺伝子変異を確認する方が確実であると考えられます。
KO動物作製の場合、モザイク個体からF1世代を作出する場合、複数の変異系統を樹立できる可能性がある点は、課題というよりも利点と考えることができます。
- 課題2. Off-target効果
- CRISPR/Casシステム利用時に特に注意すべき点ですが、gRNAが認識する塩基数が20塩基程度と短いため、標的配列と類似した配列を切断してしまうことがあります。これがOff-target効果と呼ばれるものです。その対策として、CRISPRdirectなどのガイドRNA設計ソフトウエアを用いてできるだけ類似配列の少ない標的配列を選択することや、野生型個体との交配によるOff-target siteの野生型化が現実的な解決策になると考えられる。さらには、Cas9 nickaseと2つのgRNAを用いることにより、認識配列を40塩基に増やすことで、Off-target効果を抑えるという方法も報告されています。
KACでの遺伝子改変動物作製例
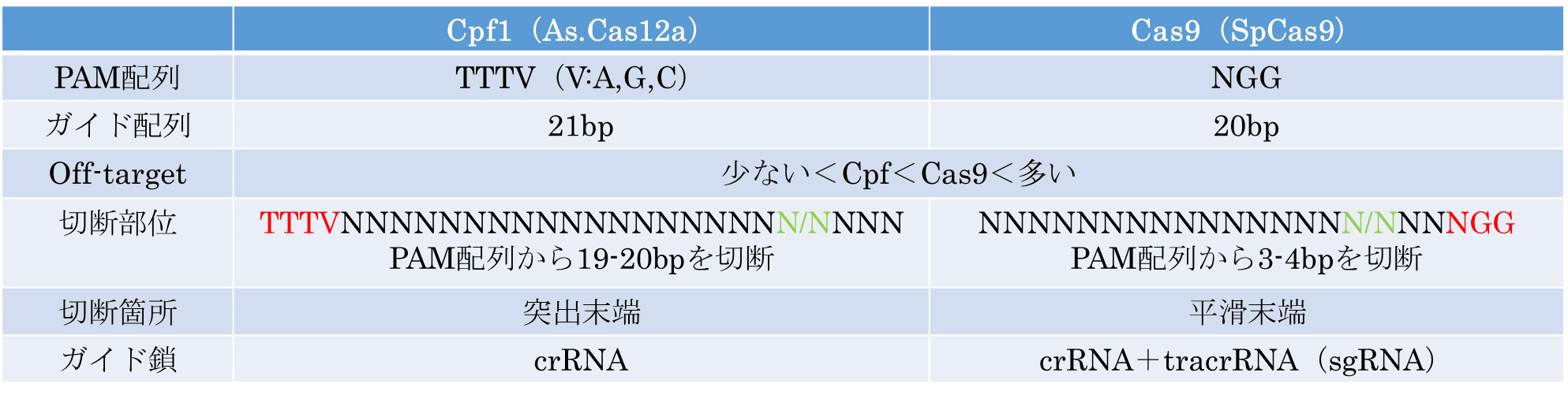
株式会社ケー・エー・シーは、米国Broad Institute社より、同社の有するCpf1(Cas12a)技術に関する特許(ならびに、特許出願)について、日本における非独占的実施権の許諾を受けており、Cpf1を用いて遺伝子改変動物作製の受託を実施しております。
V型CRISPR-Casシステムに分類されるCRISPR-Cpf1は,Ⅱ型CRISPR-Casシステムに分類されるCRISPR-Cas9と同様に、guide RNAとnucleaseとの組み合わせからなりますが、guide RNAにtracrRNAを含みません。さらにCas9のPAM配列がNGGであることに対し、Cpf1のPAM配列はTTTVである点も留意したい相違点です。Cas9のDNA切断末端が平滑末端になるのに対し,Cpf1は突出末端となる点においても異なります。Cas9とCas12aの比較は以下になります。
- Tyrosinase ノックアウトマウス作製
- Tyrosinase遺伝子は毛色決定を司る遺伝子であり、両アリルともに欠損するとアルビノ個体になることが知られています。ノックアウトの状態が毛色に反映されることから、目視で遺伝子変異の有無を確認できるため、Tyrosinaseノックアウトマウスの作製を試みました。
C57BL/6Jマウス(有色)の前核期胚を採取し、Tyrosinase遺伝子を標的としたcrRNAおよびCpf1 タンパク質をインジェクションして、偽妊娠マウスに移植しました。
その結果、21匹の産仔(F0)を得ました。そのうち16匹(76%)にアルビノ個体を含む毛色の変異が見られました。以下がF0個体の毛色の結果になります。

さらに得られたF0個体を次世代シークエンサーでの塩基配列解析を実施した結果、多くの変異を確認することができました。以下に変異の一例を示します。

導入された変異としては、5~10数bpの欠損個体が確認できました。1個体あたり数種類の変異(モザイク)が見られ、F0個体で変異導入がほぼ100%の個体も得られることを確認しています。
以上の結果から、Cas9と同様にノックアウトマウスの作製が可能であることが確認できました。ラットにおいても同様にゲノム編集できることを確認しております。
現在、cpf1を用いた遺伝子改変動物の作製は数塩基欠損のノックアウトマウスのみならず、2箇所を切断しての大規模欠損や900bp程度の目的配列を導入したノックイン動物の作製にも成功しています。
今後はflox動物、Cre挿入動物の作製の検討を行っていく予定です。